磷脂酰肌醇-3-激酶α(PI3Kα)的过度活化是多种实体瘤存活、增殖和生长的关键途径。PI3Kα作为抗肿瘤治疗的重要靶点,其高选择性抑制剂的发现成为抗肿瘤药物研发的热点,也是难点。Alpelisib是全球首个,也是唯一上市的PI3Kα选择性抑制剂,而我国尚无上市的自主知识产权的PI3Kα选择性抑制剂。PI3Kα选择性抑制剂的发现极富挑战性,首先I类PI3K四个亚型(PI3Kα、β、δ、γ)的序列同源性和结构保守性高,高度选择性抑制PI3Kα难于实现。其次,PI3Kα作为热门靶点,欧美制药公司已报道多种骨架结构的PI3Kα抑制剂,发现创新骨架并获得知识产权更是困难重重。
药学院李义平教授团队和佛罗里达大学药学院李成龙教授长期开展国际合作,致力于创新药物的发现。针对PI3Kα选择性抑制剂的发现,历经十年,在设计合成的600多个6H-苯并[c]苯并吡喃母核化合物中,发现了全新结构、具有完全自主知识产权的PI3Kα选择性抑制剂,编号为XJTU-L453。

XJTU-L453对PI3Kα具有高度选择性抑制作用,抑制活性和选择性均高于诺华已上市的药物alpelisib。通过分子动力学模拟,发现XJTU-L453的3位侧链与非保守氨基酸Gln859以及9位嘧啶环上的乙氨基与疏水核糖口袋的相互作用是其具有高度选择性的分子机制。在细胞水平上,证实XJTU-L453对PI3Kα突变型乳腺癌细胞T-47D(H1047R)和MCF-7(E545K)具有显著的增殖抑制作用,抑制活性强于alpelisib。在机制上,证实XJTU-L453抑制PI3K/AKT/mTOR信号通路,阻滞细胞周期到G0/G1期,并诱导细胞凋亡。为进一步观察XJTU-L453的体内作用,在大鼠药动学研究中,发现XJTU-L453药动学性质良好,吸收迅速,消除较快,Cmax、AUC0-t与剂量具有较好的线性关系,生物利用度为37%-41%。在荷瘤(MCF-7)重度免疫缺陷NCG鼠模型中,证实XJTU-L453具有明显的体内抗肿瘤作用,并在给药期间,各组NCG鼠体重缓慢增加,体重无明显差别,提示XJTU-L453安全性较好。研究发现了新结构类型的PI3Kα选择性抑制剂,证实了XJTU-L453是高选择性PI3Kα抑制剂,具有显著的体内外活性和良好的药动学性质,为开发自主知识产权的抗肿瘤创新药物提供了强力支撑。目前,团队已启动了XJTU-L453的临床前研究工作。

XJTU-L453与PI3Kα催化活性区(PDB ID: 5DXT)的结合模式
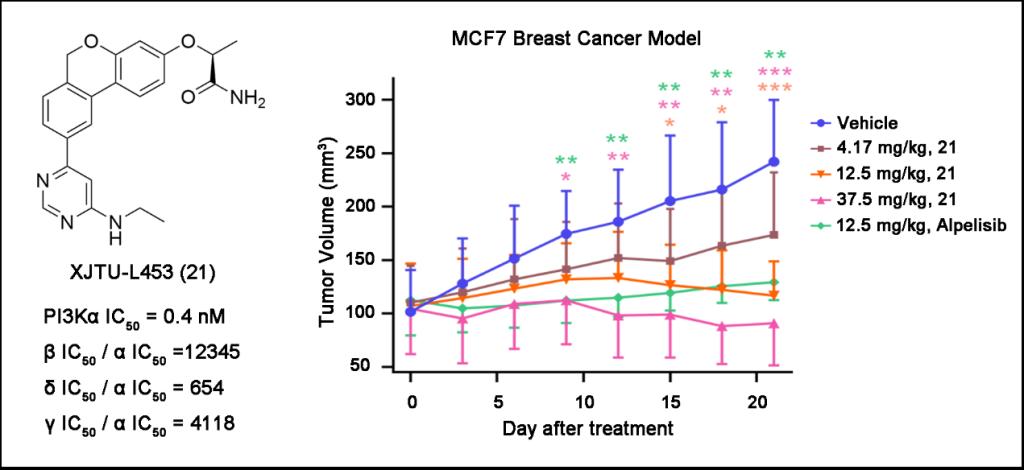
XJTU-L453对荷瘤(MCF-7)重度免疫缺陷NCG鼠的体内抗乳腺癌作用
XJTU-L453的发现工作近日以《新型6H-苯并[c]苯并吡喃系列PI3Kα选择性抑制剂的合理设计》(The Rational Design of a Novel 6H-Benzo[c]chromen Series as Selective PI3Kα Inhibitors)为题在《药物化学杂志》(Journalof Medicinal Chemistry)发表。药学院硕士生冯恒和第一附属医院生物样本信息资源中心师雪为共同第一作者,药学院李义平教授和佛罗里达大学药学院李成龙教授为通讯作者。该项研究得到了国家自然科学基金和陕西省科技计划项目的支持。
论文链接:https://doi.org/10.1021/acs.jmedchem.4c00992

 交大主页
交大主页 医学部主页
医学部主页 院长信箱
院长信箱 书记信箱
书记信箱

 官方微信
官方微信